Mercados genéricos europeos: enfoques regulatorios en la UE en 2025

En la Unión Europea, los medicamentos genéricos son la columna vertebral del sistema de salud pública. Aunque representan el 65% de todas las recetas emitidas, solo generan el 18% del valor total del mercado. Esta disparidad no es casual: detrás de cada pastilla barata hay un sistema regulatorio complejo, fragmentado y en plena transformación. En 2025, tras décadas de ajustes, la UE implementó su reforma más profunda desde 2004: el Pharma Package. ¿Qué significa esto para las empresas que fabrican genéricos? ¿Y para los pacientes que los necesitan?
Los cuatro caminos para aprobar un genérico en la UE
No existe una única forma de obtener autorización para vender un medicamento genérico en la UE. Hay cuatro vías, cada una con sus reglas, tiempos y costos. Elegir la incorrecta puede retrasar un lanzamiento por meses, o incluso años.La vía más directa es el Procedimiento Centralizado. Con una sola solicitud a la Agencia Europea de Medicamentos (EMA), el genérico obtiene autorización en los 27 países de la UE más Islandia, Liechtenstein y Noruega. Su ventaja es clara: acceso simultáneo a todo el mercado. Pero su costo es alto: entre 425.000 y 2 millones de euros en tarifas y consultoría. Solo lo usan las grandes empresas, como Sandoz, que lanzaron su versión genérica de Cosentyx en toda Europa en el segundo trimestre de 2025, 11 meses antes de lo que habría sido posible con otros métodos.
El Procedimiento de Reconocimiento Mutuo (MRP) es el más usado: el 42% de las solicitudes lo utilizan. Una empresa primero obtiene la autorización en un país (el Estado miembro de referencia), luego solicita que otros países la reconozcan. Su desventaja: cada país puede pedir ajustes. Teva experimentó esto con su genérico de rosuvastatina: aprobado en Alemania, pero retrasado 8,2 meses en Países Bajos y Bélgica por negociaciones de precios locales. El proceso, que debería durar 90 días, toma en promedio 133 días.
El Procedimiento Descentralizado (DCP) permite presentar la solicitud en varios países al mismo tiempo. Aparentemente, es más rápido. Pero en la práctica, el 37% de los casos sufren retrasos de más de seis meses. ¿Por qué? Porque países como Polonia o Rumanía interpretan las normas de calidad de forma distinta a Alemania o Francia. Un estudio de la GMDP Academy en 2024 mostró que los problemas más frecuentes están en los requisitos de estabilidad y en la caracterización de compuestos polimórficos.
Y luego está el Procedimiento Nacional, usado solo por el 5% de los fabricantes. Se trata de pedir autorización en un solo país. Es útil si quieres entrar solo en un mercado con altos reembolsos, como Francia. Pero pierdes la ventaja de la armonización. Accord Healthcare tardó 197 días en Francia, mientras que con MRP logró autorización en cinco países en 142 días.
Lo que todos deben demostrar: bioequivalencia
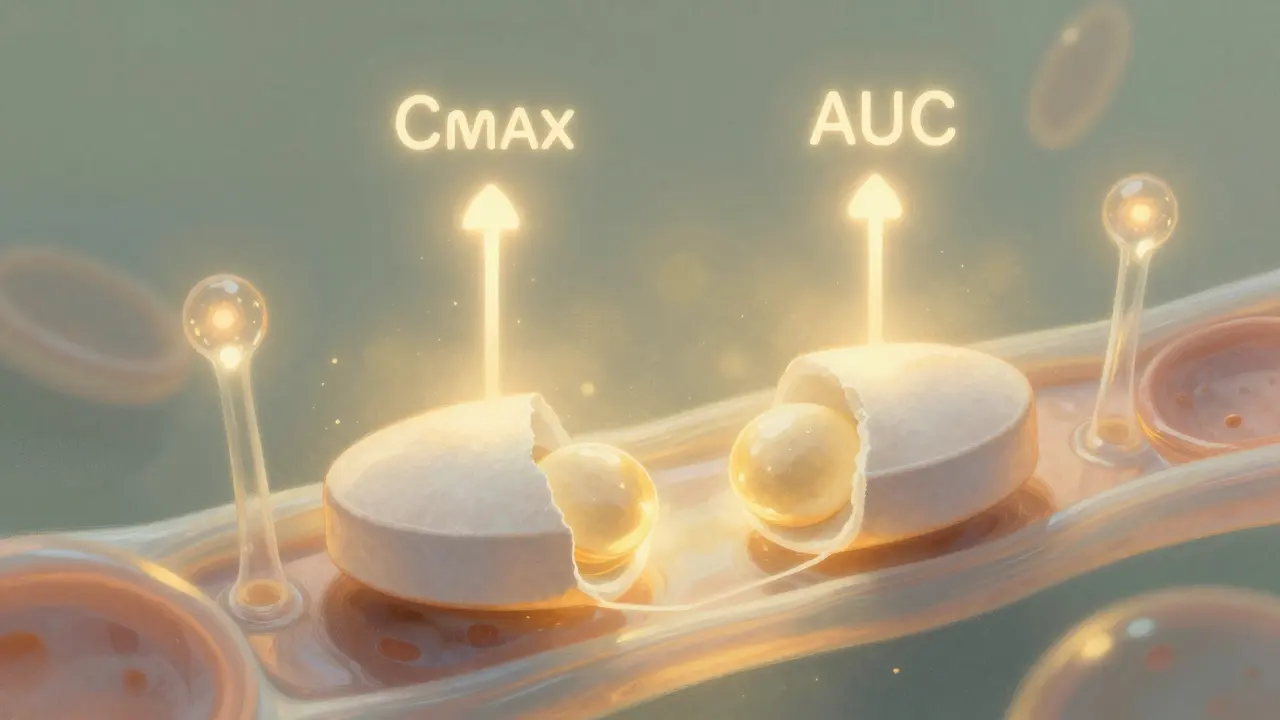
Independientemente de la vía elegida, todos los genéricos deben probar lo mismo: que funcionan igual que el medicamento original. No basta con tener la misma fórmula. Hay que demostrar que el cuerpo lo absorbe de la misma manera.La EMA exige estudios de bioequivalencia con intervalos de confianza del 90% para dos parámetros clave: la concentración máxima en sangre (Cmax) y la exposición total (AUC). Ambos deben estar entre el 80% y el 125% del producto de referencia. Es un estándar estricto. Pero algunos países lo hacen aún más difícil. El BfArM de Alemania, por ejemplo, exige estudios adicionales de farmacodinamia para inhaladores, algo que no pide la EMA. Esto crea un laberinto: un genérico puede pasar en España y fracasar en Alemania por un requisito que no es europeo, sino nacional.
Además, los estudios deben seguir la Guía de la EMA de 1998, actualizada en 2025. Esto significa más pruebas, más muestras, más tiempo. Las empresas necesitan entre 6 y 8 meses solo para diseñar y ejecutar estos estudios antes de presentar la solicitud. Y si la EMA pide más datos, el reloj se reinicia.

La reforma de 2025: el Pharma Package y sus consecuencias
La reforma más importante en 20 años entró en vigor en junio de 2025. Cambia tres reglas clave que afectan directamente cuándo y cómo entran los genéricos al mercado.Primero, la protección de datos se reduce de 10 a 8 años, con un año adicional de protección de mercado. Eso significa que, en teoría, los genéricos pueden salir antes. Pero hay un giro: si el medicamento original cumple ciertos objetivos de salud pública, ese año extra puede extenderse hasta dos años. Esto beneficia a los grandes laboratorios que invierten en tratamientos para enfermedades raras, pero puede dejar a las medianas empresas en desventaja.
Segundo, la exención Bolar se amplía. Antes, las empresas podían empezar a negociar precios y reembolsos solo dos meses antes de que expirara la patente. Ahora, pueden hacerlo seis meses antes. Esto es un cambio gigante. Permite a los sistemas de salud preparar contratos y a los genéricos presentar sus propuestas de precios antes de que el original salga del mercado. Según un modelo de REMAP Consulting, esto acelerará el lanzamiento en 4,3 meses en promedio. Pero también presiona los precios: al haber más competencia antes del lanzamiento, los precios pueden caer entre un 12% y un 18%.
Tercero, se impone la obligación de suministro. Las empresas que producen medicamentos esenciales deben garantizar que hay suficientes existencias. ¿Qué significa "suficiente"? Eso lo deciden los países, y aquí está el problema. Algunos, como Alemania, lo interpretan como mantener inventarios estratégicos. Otros, como algunos países del Este, lo ven como un requisito mínimo. Profesor Panos Kanavos, de la LSE, advierte que esta ambigüedad puede crear escasez artificial en mercados pequeños, justo donde más se necesitan los genéricos.
Quién domina el mercado y cómo
En 2024, los fabricantes indios obtuvieron el 38% de las aprobaciones de genéricos en la UE. Un aumento del 9% en solo cuatro años. Sus ventajas: costos de producción bajos, experiencia en procesos de alta volumen, y una fuerte presencia en mercados con precios más bajos como Polonia o Hungría.Pero las empresas europeas no se rinden. Sandoz y Viatris mantienen el 52% del mercado. ¿Cómo? Usan el Procedimiento Centralizado. Lanzan productos de alto valor en todo el bloque de una sola vez. No se dejan arrastrar por las lentitudes nacionales. También invierten en tecnología: en 2026, todos los productos deberán presentar su información electrónica (ePI) en formato XML. Eso requiere inversiones de entre 180.000 y 250.000 euros por empresa. Solo las grandes pueden permitírselo.
El mercado europeo de genéricos valía 42.700 millones de euros en 2024. Creció un 6,2% respecto al año anterior. Pero la brecha con Estados Unidos sigue siendo enorme: 22,4 meses entre el lanzamiento en EE.UU. y en la UE. En Canadá, esa brecha es de solo 8,7 meses. La reforma de 2025 busca reducirla. Si tiene éxito, en 2028 los genéricos podrían representar el 69,2% de las recetas, frente al 65% actual.
Desafíos reales que enfrentan las empresas
Las empresas no luchan solo contra la burocracia. Luchan contra la inconsistencia.Una encuesta de la ABPI en 2025 mostró que el 68% de las empresas consideran que los requisitos de bioequivalencia varían demasiado entre países. Un producto aprobado en Italia puede ser rechazado en España por un detalle en la formulación. El reloj regulatorio se reinicia. Cada objeción nacional vuelve a contar los 180 días de evaluación. Accord Healthcare lo vivió en carne propia: un retraso de tres meses por una solicitud de datos de estabilidad en Alemania, cuando ya habían aprobado el producto en Francia y Portugal.
Además, las autoridades nacionales no siempre siguen las pautas de la EMA. Un estudio de 2025 reveló que el 58% de las empresas recibieron respuestas contradictorias de las autoridades nacionales sobre los límites de impurezas en medicamentos con productos de referencia antiguos. ¿Qué hacer? Muchas contratan consultoras locales en cada país. Es caro. Pero más caro aún es lanzar un producto y tener que retirarlo por una norma no cumplida.
¿Qué sigue? El futuro de los genéricos en la UE
La reforma de 2025 no es el final. Es el principio de una nueva etapa. El 1 de julio de 2026 entrará en vigor el nuevo sistema de protección de datos. Se espera que acelere la entrada de genéricos para 78 biológicos en desarrollo.El Acta de Medicamentos Esenciales, aprobada en marzo de 2025, obliga a mantener reservas estratégicas de 200 genéricos críticos. Esto reducirá las escaseces, pero también añadirá capas de verificación de calidad. Las empresas tendrán que invertir más en controles y trazabilidad.
Y hay un factor externo: el Acuerdo Marco UE-EE.UU. de septiembre de 2025. Afectará los aranceles sobre ingredientes farmacéuticos. No se sabe aún cómo impactará en los costos de producción. Pero si suben, las empresas indias podrían perder ventaja, y las europeas podrían recuperar terreno.
El sistema actual es un equilibrio inestable. Por un lado, busca fomentar la competencia y bajar precios. Por otro, protege la innovación y la seguridad. La reforma de 2025 intenta corregir sus fallas. Pero hasta que todos los países actúen con la misma velocidad y claridad, el mercado seguirá siendo un rompecabezas. Para las empresas, la clave no es solo tener un buen producto. Es entender qué vía elegir, qué país seguir, y cómo navegar entre las reglas que cambian cada día.
¿Cuál es la vía más rápida para lanzar un genérico en la UE?
La vía más rápida es el Procedimiento Centralizado, pero solo si el producto tiene un potencial de ventas superior a 250 millones de euros anuales. Para productos de menor valor, el Procedimiento de Reconocimiento Mutuo (MRP) puede ser más eficiente si se elige un Estado miembro de referencia con procesos ágiles, como los Países Bajos o Suecia. Sin embargo, en la práctica, el MRP suele tardar más de lo previsto por retrasos nacionales. La única forma de acelerar realmente el lanzamiento es aprovechar la exención Bolar ampliada, que permite iniciar negociaciones de reembolso seis meses antes de la expiración de la patente.
¿Por qué los genéricos tardan tanto en llegar a algunos países de la UE?
La causa principal es la fragmentación regulatoria. Aunque la UE tiene normas comunes, cada país interpreta y aplica las reglas de forma distinta. Alemania exige pruebas adicionales para ciertos compuestos, Francia requiere documentación pediátrica específica, y países del Este tienen estándares de calidad más laxos o menos claros. Además, las negociaciones de precios y reembolso son nacionales y pueden demorar meses, incluso después de que la autorización técnica ya esté aprobada. El retraso promedio entre la aprobación técnica y el lanzamiento real es de 11,3 meses.
¿Qué cambió con la reforma del Pharma Package en 2025?
Tres cambios clave: 1) La protección de datos se redujo de 10 a 8 años, con un año extra de protección de mercado (extendible a 2 años); 2) La exención Bolar se amplió de 2 a 6 meses antes de la expiración de la patente, permitiendo negociar precios antes del lanzamiento; 3) Se impuso la obligación de suministro para medicamentos esenciales, con el objetivo de evitar escaseces. Estos cambios buscan acelerar la entrada de genéricos, reducir precios y mejorar el acceso, pero también crean nuevos desafíos para empresas medianas y en mercados pequeños.
¿Es más barato producir genéricos en la UE o en la India?
La producción es mucho más barata en la India, donde los costos laborales y regulatorios son menores. Por eso, las empresas indias controlan el 38% de las aprobaciones en la UE. Sin embargo, el costo total de entrar al mercado europeo no es solo de producción. Incluye estudios de bioequivalencia, consultoría regulatoria, y adaptación a normas nacionales. Por eso, muchas empresas indias colaboran con socios europeos para manejar los trámites. En algunos casos, el costo total de entrada puede ser comparable al de una empresa europea, especialmente si se usa el Procedimiento Centralizado.
¿Qué pasa si un genérico es aprobado en un país pero rechazado en otro?
Si se usa el Procedimiento de Reconocimiento Mutuo o el Descentralizado, un rechazo en un país puede detener todo el proceso. La autoridad que rechaza el producto puede solicitar datos adicionales, y eso reinicia el reloj regulatorio en todos los países involucrados. Esto puede sumar meses o incluso años al tiempo de lanzamiento. La única forma de evitarlo es preparar la solicitud con un conocimiento profundo de las exigencias nacionales, o usar el Procedimiento Centralizado, donde la decisión es única y vinculante para todos los países.


Ana Barić
diciembre 20, 2025 AT 21:48Me encanta que por fin se esté hablando de esto con claridad. Los genéricos son el salvavidas de muchos, y si la UE realmente quiere acceso equitativo, tiene que unificar las reglas. No puede ser que un medicamento funcione en Madrid pero no en Barcelona por un detalle de polimorfos. ¡Esto es absurdo!
Isabel Garcia
diciembre 21, 2025 AT 14:32Corrigiendo: el MRP no es más eficiente en Países Bajos, es simplemente menos corrupto. Los holandeses no juegan con los plazos, y eso hace la diferencia. Pero ojo: la exención Bolar ampliada no acelera el lanzamiento, solo la negociación de precios. La aprobación técnica sigue siendo un calvario. Y sí, la EMA no es tan neutral como dicen: los grandes laboratorios tienen acceso privilegiado a los revisores.
Nahuel Gaitán
diciembre 23, 2025 AT 02:50La verdad es que todo esto es un sistema de juego de cartas con reglas que cambian cada trimestre. Bioequivalencia? Sí, pero con un 90% de IC, y luego Alemania te pide farmacodinamia adicional porque su ministerio de salud tiene un fetish por los inhaladores. Es como si te exigieran demostrar que tu coche es igual al de tu vecino... pero también que el sonido del motor sea idéntico. ¿Por qué? Nadie lo sabe. Y los indios? Están ganando porque no les importa hacer 20 versiones del mismo producto hasta que una pase. Es pura estadística.
Amaia Davila Romero
diciembre 24, 2025 AT 01:21¿Alguien se ha preguntado por qué la reforma del Pharma Package llegó justo después de que Pfizer y Novartis hicieran lobby en Bruselas? 8 años de protección en vez de 10? Eso no es para los pacientes, es para que los laboratorios sigan cobrando 10.000€ por una pastilla. Y la obligación de suministro? Es una trampa. Si te obligan a tener inventario, y luego el precio cae, tú pierdes. Ellos ganan. Esto no es salud pública, es una operación financiera disfrazada de ley.
Andrea Coba
diciembre 25, 2025 AT 08:33yo creo que lo de los genéricos es super importante y que hay que apoyarlos porque son mas baratos y ayudan a mucha gente que no puede pagar los originales... aunque a veces me preocupa que si son muy baratos tal vez no sean tan buenos? pero no se, quizás es solo mi miedo... en fin, ojalá todo se arregle pronto
Luis Hinojosa
diciembre 26, 2025 AT 09:01Me parece que la clave está en entender que el sistema no está roto, está diseñado para ser complejo. Las empresas pequeñas no pueden competir con Sandoz porque no tienen los recursos para navegar entre 27 sistemas nacionales de aprobación, cada uno con sus propios protocolos de estabilidad, sus propios estándares de impurezas y sus propios burocratas que piden documentos que no existen en ninguna guía oficial. La solución no es más regulación, es una sola autoridad europea con poder real. Pero eso no va a pasar porque los países pequeños quieren mantener su poder de negociación. Y así seguimos.
diana jahr
diciembre 27, 2025 AT 03:48la verdad es que no sabia que habia tantas vías para aprobar un generico en la ue... me sorprendio lo de la exencion bolar que ahora es de 6 meses no de 2... eso si es un cambio real... y lo de los paises del este con normas mas laxas... bueno... no me extraña... pero ojo con la calidad... yo creo que si se puede mejorar todo esto con mas transparencia y menos papeleo... por favor que alguien haga algo
José Luis Alonso Gallardo
diciembre 28, 2025 AT 13:27Leí esto con mucho cuidado y me emocioné. Por fin alguien explica esto sin jerga de consultor. Lo que más me toca es lo de las escaseces en los países pequeños. Mi abuela en Extremadura no tiene acceso a su genérico de metformina porque el distribuidor dice que "no vale la pena" enviarlo. La reforma dice que hay que garantizar suministro... pero si no hay sanciones reales, es solo un papel. Necesitamos que alguien vaya a esos pueblos y vea qué pasa. No basta con datos.
José Manuel Martín
diciembre 29, 2025 AT 06:01¡Mira, esto es como un juego de ajedrez donde cada país tiene su propia torre y las reglas cambian cada ronda! El Procedimiento Centralizado es el mate en dos: rápido, limpio, brutal. Pero solo lo usan los que tienen dinero para pagar el tablero. Los demás se quedan jugando al parchís con 27 cajas de reglas, cada una con un gato dentro. Y los indios? Son los que compran las fichas baratas y las arrojan hasta que una encaja. No es ético, no es justo, pero es inteligente. Y la reforma? Es como si alguien pusiera un cartel que dice "¡ahora hay más espacio!"... pero no cambia el tamaño del tablero. Seguimos en el mismo laberinto.
JULIO ANDINO
diciembre 30, 2025 AT 16:30¿De verdad creen que esto es una reforma? Es una pantomima. La EMA es un instrumento de los grandes laboratorios, y los "genéricos" que llegan son versiones ligeramente modificadas para evadir patentes. El 80-125% de bioequivalencia? Una burda mentira. La farmacocinética no es lineal, y los polimorfos alteran la absorción en pacientes con enfermedades crónicas. Pero nadie lo estudia porque no es rentable. Y los indios? Claro, producen a bajo costo... pero con ingredientes de dudosa procedencia. ¿Alguien ha revisado los laboratorios de Hyderabad? No, porque Bruselas prefiere la apariencia de competencia a la realidad de la seguridad. Esto es un fraude disfrazado de progreso.
Martin Dávila
enero 1, 2026 AT 07:39...y... esto... ¿es serio?... 425.000 euros... para... una pastilla...?... y luego... los países... que... no... se... ponen... de... acuerdo...?... y... los... indios... que... ganan...?... y... la... exención... Bolar...?... y... el... Pharma... Package...?... y... la... obligación... de... suministro...?... y... los... 11... meses... de... retraso...?... y... los... 68%... de... empresas... que... dicen... que... es... un... caos...?... y... los... 58%... con... impurezas... contradictorias...?... y... el... Acuerdo... UE-EE.UU...?... y... el... Acta... de... Medicamentos... Esenciales...?... y... los... 200... genéricos... críticos...?... y... los... 22,4... meses... de... brecha... con... EE.UU...?... ¿Y... qué... pasa... con... mi... abuela... que... necesita... su... metoprolol...?... ¿Alguien... me... lo... explica... en... una... frase...?... Por... favor...?... Porque... yo... ya... me... rindo...